
ポスト「京」重点課題1 ワークショップ
課題研究実施者と若手研究者を含むプロジェクト参加者を対象に研究ワークショップを年2回開催し、進捗確認とサブ課題間・分担機関間での相互連携を推進しています。第1回 ワークショップ
日時
2016年9月9日(金)10:30~16:50
場所
理化学研究所 生命システム研究センター B棟大会議室
要旨報告
■オープニングあいさつ
奥野 恭史(
重点課題1 課題責任者、理化学研究所 生命システム研究センター、京都大学大学院医学研究科)
柳田 敏雄(理化学研究所生命システム研究センター センター長)
■口頭発表
| |
1.分子動力学法を用いたタンパク質ー薬剤複合体の高精度予測 サブ課題A 尾嶋 拓(理化学研究所 生命システム研究センター) |
| 我々は、薬剤開発加速のために、分子動力学(MD)アプリケーション「GENESIS」を用いたタンパク質—薬剤の結合構造と結合自由エネルギー(BFE)の高精度予測を目指している。本発表では、液体論を用いたアクチンーミオシンのBFE計算法を紹介し、GENESISによるリガンド結合MD計算を報告した。長時間MDではリガンド結合の観測は難しく、結合プロセスや結合の阻害要因を解析し、結合を加速する手法が必要であることがわかった。AMPAレセプターの変異の影響はX線結晶構造では説明できず、動的な振る舞いの観測が必要である。MDシステムのセットアップと力場を開発し、自由エネルギー計算によるメカニズムの解明を現在行っている。 | |
【図】GENESISによるリガンド結合MD |
 |
2.エネルギー表示溶液理論を用いた蛋白質複合体構造予測 サブ課題A 竹村 和浩(東京大学 分子細胞生物学研究所) |
| 剛体ドッキングとクラスタリング/リランキングプログラムであるCyClusを組み合わせて正解構造を含む100-300の蛋白質-蛋白質複合体モデルを生成し、エネルギー表示法を用いて複合体モデル間の結合自由エネルギーの計算を行った。複合体界面の蛋白質間の相互作用や界面に存在する水分子等も含め正しい構造を作れている場合は、高精度で複合体モデルの評価を行うことができることを示した。 | |
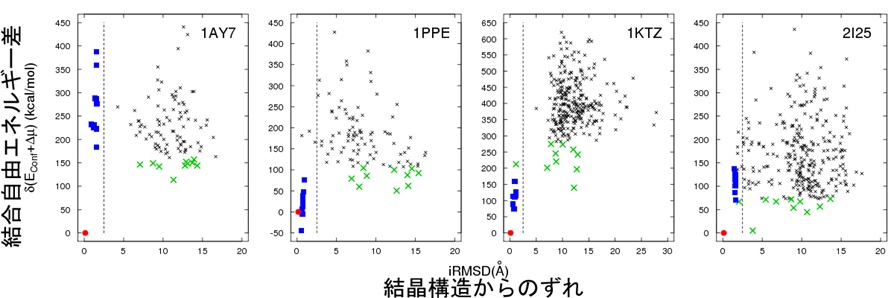 【図】蛋白質-蛋白質複合体の結合自由エネルギー差計算例 エネルギー表示溶液理論を用いて蛋白質-蛋白質複合体の結合自由エネルギー差を計算した例、高精度で正しい複合体モデルを再安定として評価できる。 |
 |
3.タンパク質と薬剤分子のQM/MM RWFE-SCF法による結合自由エネルギー計算の高精度化 サブ課題A 長谷川 太祐(京都大学理学研究科 化学専攻 理論化学研究室) |
| QM/MM RWFE-SCF法によるタンパク質と薬剤分子の結合自由エネルギー計算手法について報告した。従来のQM/MM RWFE-SCF法を拡張し、多極子ESP演算子とQM領域の剛体MD化により、タンパク質と薬剤分子の静電相互作用の記述の高精度化と薬剤分子の並進・回転の効率的な探索が可能となった。また改良したQM/MM RWFE-SCF法を応用してHen egg white lysozymeのE35のpKa計算を行ったことを報告した。 | |
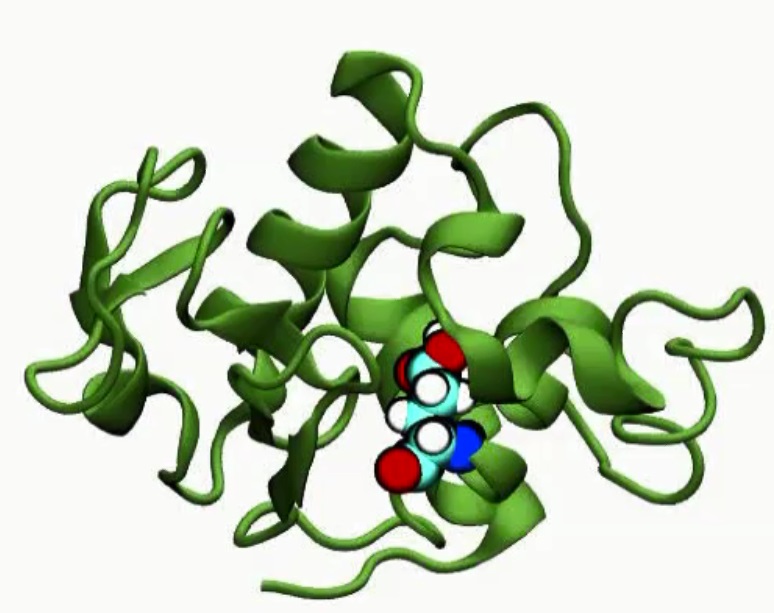 |
【図】拡張したQM/MM RWFE-SCF法で計算した HEWLのMDスナップショット
|
 |
4.蛋白質・DNA複合系の高精度粗視化モデリングに向けて サブ課題A 高田 彰二(京都大学大学院理学研究科) |
| 我々が進めている粗視化分子シミュレーションの技術的進捗と、細胞核分子モデリング への応用について報告する。まず、粗視化シミュレーションソフトウエアCafeMolのバージ ョン3.0を8月末に一般公開した。今回、新しいDNAモデルが実装され、従来よりかなり高精度で、配列依存的なDNA物性を再現できるようになった。粗視化モデルから全原子モデルを再構築するツールも概ね完成してきた。また、Protein Binding Microarray等の蛋白質DNA相互作用の高スループット実験データを分子シミュレーションに取り込む方法を開発した。応用計算では、ヌクレオソームと転写因子の競合過程等において興味深い現象をシミュレーション中で見出している。 | |
 |
粗視化分子シミュレーションソフトウエアCafeMol 3.0を2016年8月31日に一般公開した。
 |
5.ビタミンD受容体リガンド結合ドメインのMD-SAXS相関構造解析 サブ課題B 浴本 亨(横浜市立大学 生命医科学研究科) |
| 核内受容体ビタミンD受容体は、骨粗鬆症等の疾病に関係する創薬ターゲットである。X線小角散乱(SAXS)と分子動力学計算(MD)を組み合わせた相関構造解析により、アポ体とアンタゴニスト複合体の溶液構造を原子レベルで明らかにした。動的分子機能制御を目指し、「京」を使った大規模MDから、新たに提案した2つのへリックスの構造変化が連動するリガンド結合機構の検証や、活性と構造の関係の理解等に取り組んでいきたい。 | |
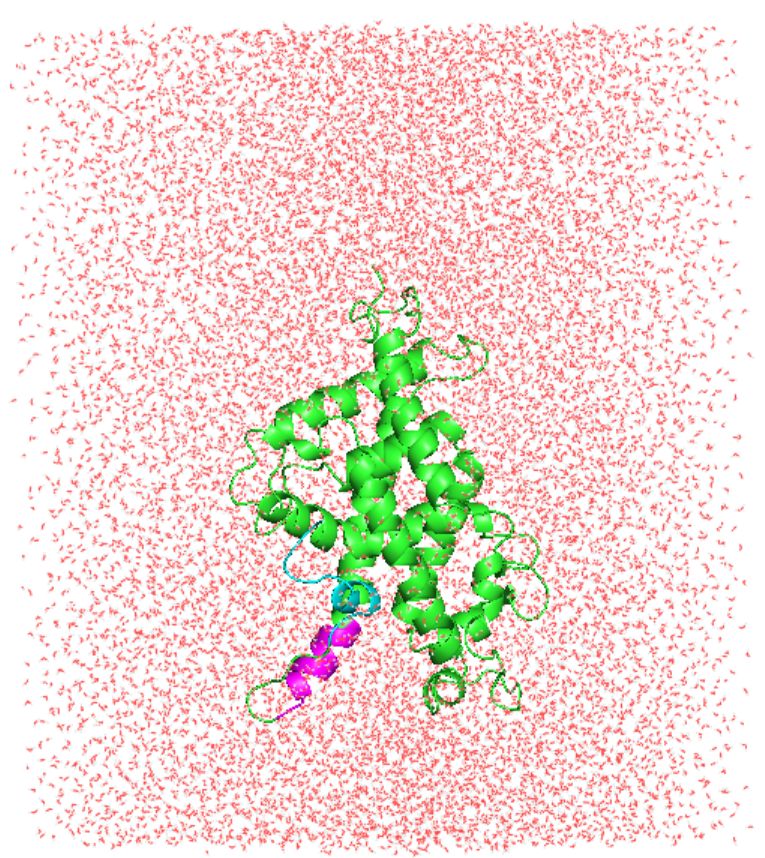 |
【図】アポ体の溶液構造モデル MDから明らかになった、SAXS実験プロファイルと一致した溶液構造モデル |
 |
6.HBVの分子論的研究 サブ課題B 藤本 和士(名古屋大学大学院工学研究科) |
| 我々は、分子動力学(MD)計算ソフトMODYLASを用いて、B型肝炎ウィルス(HBV)と薬剤の親和性を明らかにすることを目指している。本発表では、HBVおよびそのRNAのモデリング、RNA複製阻害剤(エンテカビル)のポテンシャル開発について報告した。さらに、形成促進剤がHBVの構造変化を誘起させ、構造を安定化することも報告した。 | |
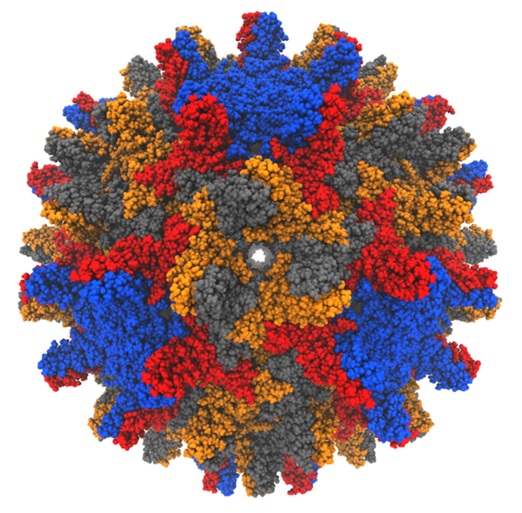 |
【図】B型肝炎ウィルス B型肝炎ウィルスは1種類のタンパク質から構成されており、3回回転対称軸上に大きなトンネルが見られる。 |
 |
7.抗原抗体のMD計算と界面構造の解析 サブ課題B 山下 雄史(東京大学先端科学技術研究センター) |
| 本発表では、タンパク質間相互作用を理解するために開発してきた計算・解析手法を議論する。まず、抗原抗体複合体の解離に対する平均力ポテンシャルを安定して計算するために、multi-step targeted MD(mTMD)法を開発した。本手法により、タンパク質の内部構造に歪みが生じないため、解離の自由エネルギーの過大評価を抑えることができるようになった。また、タンパク質の重要であるが微小な構造変化を検出するために、ラマチャンドラン角のdirectional analysisを開発した。この方法により、重要だが微小な構造変化を検出できるようになった。 | |
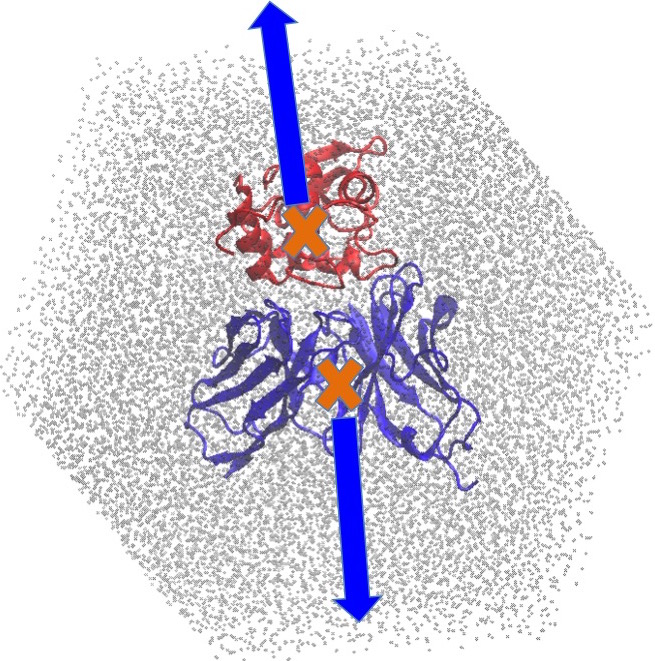 |
【図】抗原抗体の複合体 グレーの点は水分子、オレンジのリボンは抗原、シアンのリボンは抗体を表す。矢印は解離方向を表す。 |
 |
8.ヒストンの翻訳後修飾がヌクレオソーム構造に与える影響の解析 〜分子動力学シミュレーションによる研究〜 サブ課題B 池部 仁善(量子科学技術研究開発機構 生体分子シミュレーショングループ) |
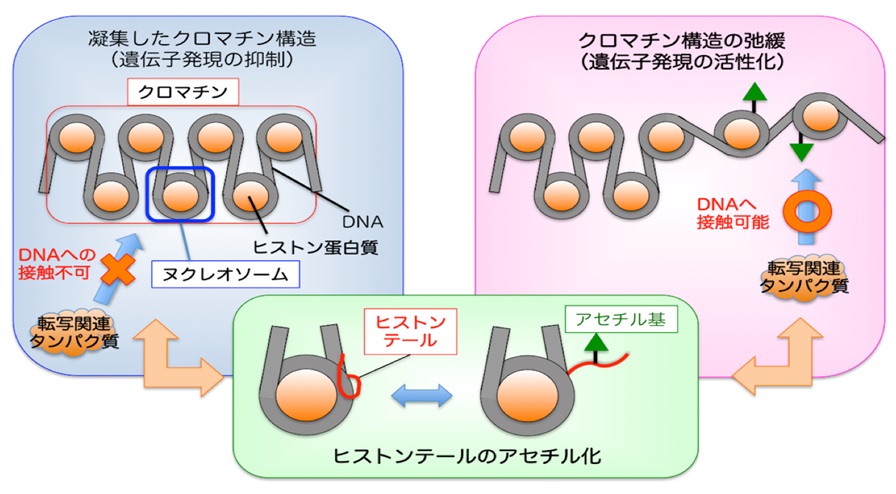
| 核内DNA-タンパク質複合体(ヌクレオソーム)を構成するヒストンタンパク質の末端領域、ヒストンテールへの翻訳後修飾は、転写などのDNA機能制御において重要な役割を果たしている。本研究では、分子動力学シミュレーションの一種であるALSD法を用い、翻訳後修飾(アセチル化)の数、修飾パターンの違いが、ヒストンテールやヌクレオソームの構造に与える影響について解析した結果を報告する。 | |
【図】アセチル化によるヌクレオソームの構造変化 |
 |
9.タンパク質-化合物結合構造を高精度に予測する方法論の開発 サブ課題C 荒木 望嗣(理化学研究所 計算科学研究機構) |
| ポスト「京」サブ課題Cで開発する創薬ビッグデータ統合システムは複数の創薬計算技術から構成される。H27年度は、システムの中核となる、タンパク質-薬剤結合構造を正確に予測するための方法論を開発した。CDK2キナーゼと結合構造既知の低分子化合物群を使用して本法の予測精度を検証したところ、従来法と比べて大きく予測精度が向上した。今後は計算パラメータを精密化することにより、幅広いタンパク質に対して頑強に予測できる方法論に改良する予定である。 | |
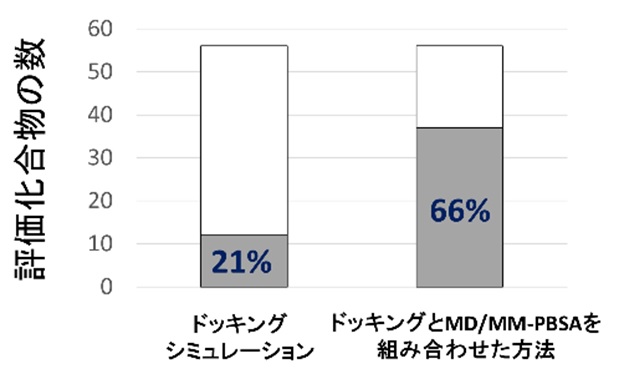 |
【図】タンパク質-化合物結合構造の予測正答率 検証には、CDK2キナーゼと共結晶構造が明らかになっているATP結合阻害剤56種を 使用した。ドッキングシミュレーションのみによる予測正答率は21%(12化合物)にとどまったが、MD/MM-PBSAと組み合わせることにより、66%(37化合物)にまで上昇した。 |
 |
10.アンサンブル構造を用いたリガンド結合部位予測 サブ課題C 広川 貴次(産業技術総合研究所創薬分子プロファイリング研究センター) |
| 本研究では、化合物の結合に伴う構造変化を想定して、アポ体を初期構造に、分子動力学計算を用いてタンパク質の揺らぎのコンフォメーションを発生させ、探索や設計の目的に応じて揺らぎのコンフォメーションを選定するシステムの開発を試みた。最初に、分子動力学計算によってトラジェクトリーを生成し、複数のトラジェクトリー構造上において、化合物の結合ポケット周辺の形状を偽原子で予測する。続いて、偽原子の存在密度を解析し、目的の化合物の設計指針に最適な構造を、偽原子からなる疑似化合物と実際の化合物等との簡易形状比較で選定する。現在、キナーゼを中心に、いくつかの標的タンパク質に対して本手法を検証中である。 | |
 |
11.Machine-Learning-Enhanced MM-PBSAの開発 サブ課題C 寺山 慧(東京大学大学院新領域創成科学研究科メディカル情報生命専攻) |
| バーチャルスクリーニングによって医薬品候補を予測するためには, タンパク質と候補化合物の間に働く結合自由エネルギーを正確に計算する必要がある. MM-PBSA法は比較的高い精度で結合自由エネルギーを推定できる手法であるものの, 計算コストが高い. 本発表では, 一定の状況下において機械学習の一手法である最適腕選択アルゴリズムを用いることで, MM-PBSA法の計算コストが削減できることを実験的に示した。 | |
 |
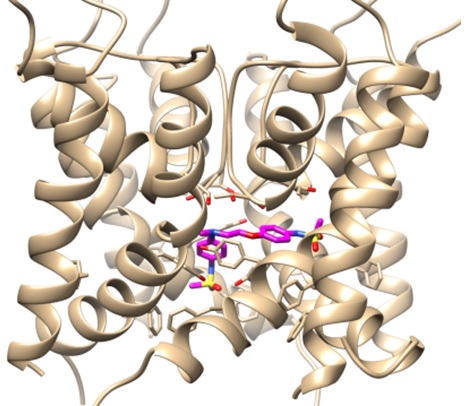
12.電位依存性イオンチャネルのモデリングと薬剤-チャネル相互作用の解析 サブ課題C 根上 樹(東京大学大学院農学生命科学研究科) |
| 心筋における電位依存性チャネルと薬剤の相互作用予測の準備として、イオンチャネルのモデリングおよびチャネル構造の評価を行った。ホモロジーモデリングによるhERGチャネルのporeモデル構造と8種類の薬剤とのドッキングシミュレーションを実行したところ、相互作用残基および結合親和性の傾向について実験と整合する結果が得られた。また、分子動力学計算において、Kv1.2チャネルのS4-S5 linkerからS6 helixの末端までのpore部分構造が安定であることが示された。 | |
|
【図】GENESISによるリガンド結合MD Glideドッキングシミュレーションにより得られた、hERGチャネルのporeモデル(茶)とdofetilide(マゼンタ)のドッキング構造 |
■クロージング
講評、まとめ、今後の予定
奥野 恭史(重点課題1
課題責任者、理化学研究所 生命システム研究センター、京都大学大学院医学研究科)
集合写真

特別講演 「楽しい可視化を行うために」
【講師】株式会社サイアメント 瀬尾 拡史【要旨】HPCI戦略プログラム分野1「予測する生命科学・医療および創薬基盤」の成果の1つである「核内混み合い環境でのヌクレオソーム、クロマチンの機能発現機構」の可視化映像の制作裏話をお話します。 シミュレーションデータとハリウッド映画用の3DCGソフトウェアとの連携や、ストーリー作成のポイントなど、技術的な観点から可視化のちょっとしたテクニックまで、具体的に紹介致します。

細胞内分子ダイナミクスシミュレーション
核内混み合い環境でのヌクレオソーム、クロマチンの機能発現機構
(HPCI戦略プログラム 分野1「予測する生命科学・医療および創薬基盤」より)
